DeepExplainer Genomics Example
This runs DeepExplainer with the model trained on simulated genomic data from the DeepLIFT repo (https://github.com/kundajelab/deeplift/blob/master/examples/genomics/genomics_simulation.ipynb), using a dynamic reference (i.e. the reference varies depending on the input sequence; in this case, the reference is a collection of dinucleotide-shuffled versions of the input sequence)
The simulated data was as follows:
1/4 sequences with BOTH 1-3 instances of a GATA_disc1 motif AND 1-3 instances of a TAL1_known1 motif; these were labelled 1,1,1
1/4 sequences with 1-3 instances of a GATA_disc1 motif embedded; these were labelled 0,1,0
1/4 sequences with 1-3 instances of a TAL1_known1 motif embedded; these were labelled 0,0,1
1/4 sequences with no motif; these were labelled 0,0,0
[1]:
%matplotlib inline
Obtain data and keras model
Pull in the relevant data
[2]:
! [[ ! -f sequences.simdata.gz ]] && wget https://raw.githubusercontent.com/AvantiShri/model_storage/db919b12f750e5844402153233249bb3d24e9e9a/deeplift/genomics/sequences.simdata.gz
! [[ ! -f keras2_conv1d_record_5_model_PQzyq_modelJson.json ]] && wget https://raw.githubusercontent.com/AvantiShri/model_storage/b6e1d69/deeplift/genomics/keras2_conv1d_record_5_model_PQzyq_modelJson.json
! [[ ! -f keras2_conv1d_record_5_model_PQzyq_modelWeights.h5 ]] && wget https://raw.githubusercontent.com/AvantiShri/model_storage/b6e1d69/deeplift/genomics/keras2_conv1d_record_5_model_PQzyq_modelWeights.h5
! [[ ! -f test.txt.gz ]] && wget https://raw.githubusercontent.com/AvantiShri/model_storage/9aadb769735c60eb90f7d3d896632ac749a1bdd2/deeplift/genomics/test.txt.gz
Load the data
[3]:
! pip install simdna
Requirement already satisfied: simdna in /home/xiao/miniforge3/envs/shap/lib/python3.11/site-packages (0.4.3.2)
Requirement already satisfied: numpy>=1.9 in /home/xiao/miniforge3/envs/shap/lib/python3.11/site-packages (from simdna) (1.26.3)
Requirement already satisfied: matplotlib in /home/xiao/miniforge3/envs/shap/lib/python3.11/site-packages (from simdna) (3.8.2)
Requirement already satisfied: scipy in /home/xiao/miniforge3/envs/shap/lib/python3.11/site-packages (from simdna) (1.11.4)
Requirement already satisfied: contourpy>=1.0.1 in /home/xiao/miniforge3/envs/shap/lib/python3.11/site-packages (from matplotlib->simdna) (1.2.0)
Requirement already satisfied: cycler>=0.10 in /home/xiao/miniforge3/envs/shap/lib/python3.11/site-packages (from matplotlib->simdna) (0.12.1)
Requirement already satisfied: fonttools>=4.22.0 in /home/xiao/miniforge3/envs/shap/lib/python3.11/site-packages (from matplotlib->simdna) (4.47.2)
Requirement already satisfied: kiwisolver>=1.3.1 in /home/xiao/miniforge3/envs/shap/lib/python3.11/site-packages (from matplotlib->simdna) (1.4.5)
Requirement already satisfied: packaging>=20.0 in /home/xiao/miniforge3/envs/shap/lib/python3.11/site-packages (from matplotlib->simdna) (23.2)
Requirement already satisfied: pillow>=8 in /home/xiao/miniforge3/envs/shap/lib/python3.11/site-packages (from matplotlib->simdna) (10.2.0)
Requirement already satisfied: pyparsing>=2.3.1 in /home/xiao/miniforge3/envs/shap/lib/python3.11/site-packages (from matplotlib->simdna) (3.1.1)
Requirement already satisfied: python-dateutil>=2.7 in /home/xiao/miniforge3/envs/shap/lib/python3.11/site-packages (from matplotlib->simdna) (2.8.2)
Requirement already satisfied: six>=1.5 in /home/xiao/miniforge3/envs/shap/lib/python3.11/site-packages (from python-dateutil>=2.7->matplotlib->simdna) (1.16.0)
[4]:
import gzip
import simdna.synthetic as synthetic
data_filename = "sequences.simdata.gz"
# read in the data in the testing set
test_ids_fh = gzip.open("test.txt.gz", "rb")
ids_to_load = [x.decode("utf-8").rstrip("\n") for x in test_ids_fh]
data = synthetic.read_simdata_file(data_filename, ids_to_load=ids_to_load)
[5]:
import numpy as np
# this is set up for 1d convolutions where examples
# have dimensions (len, num_channels)
# the channel axis is the axis for one-hot encoding.
def one_hot_encode_along_channel_axis(sequence):
to_return = np.zeros((len(sequence), 4), dtype=np.int8)
seq_to_one_hot_fill_in_array(zeros_array=to_return, sequence=sequence, one_hot_axis=1)
return to_return
def seq_to_one_hot_fill_in_array(zeros_array, sequence, one_hot_axis):
assert one_hot_axis in (0, 1)
if one_hot_axis == 0:
assert zeros_array.shape[1] == len(sequence)
elif one_hot_axis == 1:
assert zeros_array.shape[0] == len(sequence)
# will mutate zeros_array
for i, char in enumerate(sequence):
if char in ("A", "a"):
char_idx = 0
elif char in ("C", "c"):
char_idx = 1
elif char in ("G", "g"):
char_idx = 2
elif char in ("T", "t"):
char_idx = 3
elif char in ("N", "n"):
continue # leave that pos as all 0's
else:
raise RuntimeError("Unsupported character: " + str(char))
if one_hot_axis == 0:
zeros_array[char_idx, i] = 1
elif one_hot_axis == 1:
zeros_array[i, char_idx] = 1
onehot_data = np.array([one_hot_encode_along_channel_axis(seq) for seq in data.sequences])
Load the model
[6]:
from keras.models import model_from_json
# load the keras model
keras_model_weights = "keras2_conv1d_record_5_model_PQzyq_modelWeights.h5"
keras_model_json = "keras2_conv1d_record_5_model_PQzyq_modelJson.json"
keras_model = model_from_json(open(keras_model_json).read())
keras_model.load_weights(keras_model_weights)
Install the deeplift package for the dinucleotide shuffling and visualization code
[7]:
!pip install deeplift
Requirement already satisfied: deeplift in /home/xiao/miniforge3/envs/shap/lib/python3.11/site-packages (0.6.13.0)
Requirement already satisfied: numpy>=1.9 in /home/xiao/miniforge3/envs/shap/lib/python3.11/site-packages (from deeplift) (1.26.3)
Compute importance scores
Define the function that generates the reference, in this case by performing a dinucleotide shuffle of the given input sequence
[8]:
from deeplift.dinuc_shuffle import dinuc_shuffle
def shuffle_several_times(s):
s = np.squeeze(s)
return dinuc_shuffle(s, num_shufs=100)
Run DeepExplainer with the dynamic reference function
[9]:
import shap
np.random.seed(1)
seqs_to_explain = onehot_data[[0, 3, 9]] # these three are positive for task 0
dinuc_shuff_explainer = shap.DeepExplainer((keras_model.input, keras_model.output[:, 0]), shuffle_several_times)
raw_shap_explanations = dinuc_shuff_explainer.shap_values(seqs_to_explain, check_additivity=False)
Visualize scores on individual sequences
[10]:
from deeplift.visualization import viz_sequence
# project the importance at each position onto the base that's actually present
dinuc_shuff_explanations = np.sum(raw_shap_explanations, axis=-1)[:, :, None] * seqs_to_explain
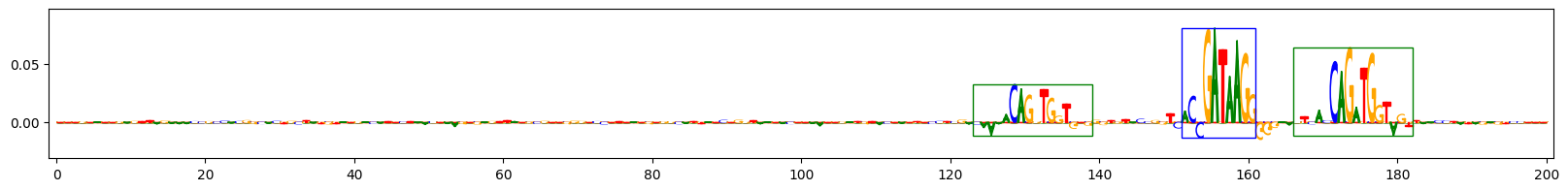
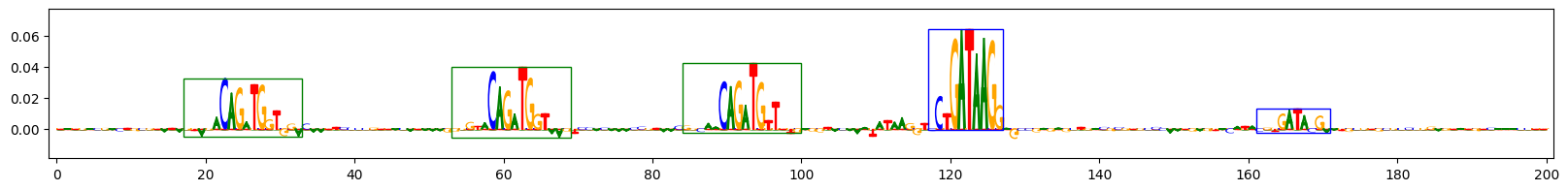
for idx, dinuc_shuff_explanation in zip([0, 3, 9], dinuc_shuff_explanations):
print("Scores for example", idx)
highlight = {
"blue": [
(embedding.startPos, embedding.startPos + len(embedding.what))
for embedding in data.embeddings[idx]
if "GATA_disc1" in embedding.what.getDescription()
],
"green": [
(embedding.startPos, embedding.startPos + len(embedding.what))
for embedding in data.embeddings[idx]
if "TAL1_known1" in embedding.what.getDescription()
],
}
viz_sequence.plot_weights(dinuc_shuff_explanation, subticks_frequency=20, highlight=highlight)
Scores for example 0
Scores for example 3
Scores for example 9
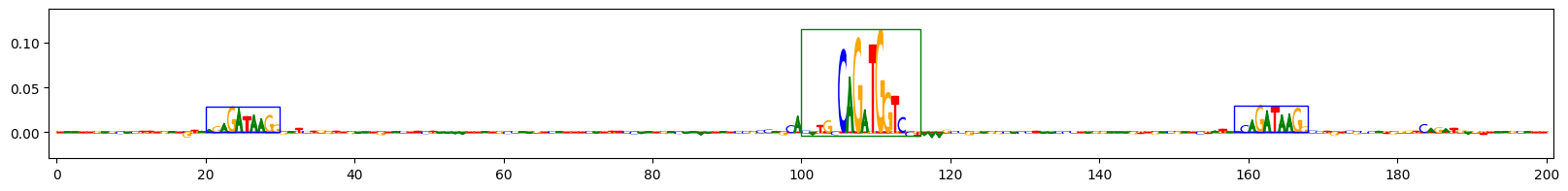
The plots above show the importance scores for three example sequences on the task that predicts sequences containing both GATA_disc1 and TAL1_known1 motifs. Letter height reflects the score. Blue boxes indicate the ground-truth locations of the inserted GATA_disc1 motifs, green boxes indicate the ground-truth locations of the inserted TAL1_known1 motifs.